演講主題: Some Computer Science
Techniques in Bioinformatics
演講者: 許聞廉,中央研究院資訊所研究員
紀錄組別: 第八組
組員: 91703001張世元 91703010 王加元
91703012 吳仕斌 91703030 江致廣
演講摘要:
生物資訊,並不像我們所熟知的一般computer science的領域,充滿了不確定性,我們不能單純以「正確」(rule-based algorithm)來進行判斷,因此,我們需要容錯的演算法來處理生物資訊方面的data → corpus-based algorithm。然而儘管這些生物資訊總是錯誤百出,computer science仍然希望能夠準確的預測。
在生物資訊當中,有許多應用到 computer science 演算法的地方,例如從較低維的蛋白質結構預測高維的結構。我們可以以一個translation problem來解釋它,也就是從低維translate到高維,最大的差異在於蛋白質的這些「word」或是「phrase」都是未經定義的,而且local一個簡單的改變可能就會影響整個高維度空間的結構。核磁共振亦是,為了解決關於蛋白質的構造,從簡單的一度空間到複雜的三度空間,一般的作法是利用NMR去猜測分子結構,把實驗的結果結合起來構成大概的蛋白質構造。
其中,也有牽涉到自然語言的部份。實做上我們可能每7個letter做一個window size,將其視為一個phrase,用自然語言模擬的方法,將knowledge base裡面進行比對。得到的結果,正確率與match rate產生正相關,比起nureral network正確率幾乎維持在同樣的水準,這也表示,當我們knowledge base的資料越多時,我們可能可以得到更精確的答案。也就是在80%以上,我們的結果有較好的參考價值。
接下來也是運用chemical shift structure來預測胺基酸構造,委員也舉一列車的例子來說明,如何以自己現有的知識來排出整列車,影射蛋白質的結構也是由一堆knowledge base來建構出來。他還提到有兩組建構重組的方法:maximum-independent set和generic algorithm;前者採用sequence間的互斥性質,後者運用block正確率大小來建構。
再來談有關到literature mining的部份,一種data mining.literature mining,如同google search,永遠層出不窮,變化無端。named entities recognition則是說,為了把一個disease描述出來,或者把一個protein作解釋,生物學家有把各種protein拿去做classification,方便查找protein和protein間的關係。比照google search文字與文字之間作關係定義,蛋白質的命名方式比這個更為複雜,至少現階段來說,還沒有一個能完全用一行文字來命名蛋白質.
而named template則運用兩種方法, phrase和dictionary based.phrase利用蛋白質作用做出來,而dictionary based則以自然法則來建構template。其他建構的方法還有利用字串組來組成template等等。
從本次演講中,我們可以概括性的看到整個生活資訊領域中的研究方法,以及會遇到的困難。
演講詳細內容:
錯誤是正常的現象:更能容錯的演算法:
我們最近這幾年做生物資訊方面,今天要談的是,我認為跟自然語言比較有關係或者是和knowledge-based有關係的這些題目。
最早,我們是從這個演算法的角度進入這個bio-informatics。演算法,各位都知道,演算法比較在computer science裡面是比較偏discrete mathematics,所以它所需要的data本身比較不能夠容忍太多的錯誤。那個時候我們遇到一個問題,就是生物資訊的data,基本上都是錯誤百出的,所以錯誤是個正常的現象。所以,通常碰到這種問題的時候,你怎麼樣能夠把以前的algorithm修改一下,像我們以前寫algorithm,就是if-then-else,這些if的assumption在生物的data裡面都不成立,因為它有error的關係。這樣的時候,你可不可以把一個這樣的演算法做某程度的改變,讓它能夠應用於有錯誤,也就是比較算是容錯的演算法上面。
下面有這樣很多的條件,是生物資訊所遇到的問題。以前在做演算法碰到有這種所謂search optimal solution的時候,就會去做approximation。但是生物的問題做approximation也不容易,因為它不是一個single objective。只有當objective是唯一的時候才可以做approximation,maximize也是,minimize也是。objective事實上是蠻多的,它比較是一種constraint satisfaction的programming而不是這個objective的optimization。所以,在這種情況我們必須做很多不同的處理。另外就是我剛剛講的error,有很多的演算法處理error是以比較global的方法,就是假設這個error case分布是平均的。但是,在生物資訊裡面,這些實驗室的error,譬如說你看micro array的Robot在點每一個cell的時候,每個地方的error比例相差非常大,所以不能用這種uniform distribution。如果一個local的noise非常嚴重,你有沒有辦法能夠事先知道,然後detect出來,所以這個是一個蠻重要的事情。另外一點很重要的就是說,以前我們design algorithm的時候就是assume你是處理arbitrary
data。在生物資訊裡面,由於我們剛才講的問題,事實上就是說像DNA在複製的時候有很多錯誤,但是naturally有個辦法能夠自動把這些錯誤修補回來,而這些錯誤發生的時空和這個背景你不太容易掌握,但是,為什麼自然界可以很容易的把這些問題解決?這個是比較有趣的。
錯誤是正常的現象:更能容錯的演算法:
事實上,在我們的看法裡,它有點像自然語言,我們在講話的時候有很多的漏字或者講的不清楚或者語意不清,但是聽的人基本上都可以restore回來,所以它很像這個DNA sequence和protein sequence,在某個層面,很像自然語言的sequence,所以它有很多容錯的能力,而這是因為它有很多structure。我們在聽自然語言也是一樣,有很多的structure可以使用,所以你不會太在乎他某一個字講的輕重,或者是不是很清楚。在做這類問題的話,就比較要學習自然語言以前的處理方法,就是你要面對的是一個real data。你不要去處理一個artificial或者是ideal的data set或者arbitrary data set。就想語音輸入,你不會處理一個沒有意義的文字,你一定會處理一個有意義的文字,然後這個裡面,你才能把一些容錯的機制建立進去。所以我們在做生物資訊的時候,也是要處理real data。
這裡簡單講一下你怎樣去catch一個data裡面的structure,在最早的時候,數學裡面比較強調這種closed-form solution,現在事實上當然是很少了。就像這個一元二次方程式的解有個很漂亮的form
solution。現在,在生物資訊裡面有很多的數學或是computer science的問題都不是這樣子有closed-form solution。在沒有closed-form solution的時候,通常它會給你一個algorithm。去解這個algorithm,本身算出來的答案可能就是你要的,但是當這個algorithm遇到這個data有error的時候呢?你有沒有辦法能夠deal with?
Rule-based algorithm 和 corpus-based
algorithm:
這個是我們現在要處理的問題。通常algorithm有兩種:if-then-else,這是我們一般寫program的algorithm,是偏向於rule-based的。以前我們做的這些discrete algorithm或各位寫的這些Java的程式基本上都是rule-based。你的rule,就寫在你的程式裡面,所以是knowledge和programming是合在一起的。那這樣有一個很大的困難,通常生物學家不會寫程式,他就沒有辦法去了解你的程式,也沒有辦法modify,所以,你必須要幫他寫的很完整,或者每次修改都要找你。這個事實上是非常不合理的。那這是rule-based algorithm一個很大的問題。
另外的是corpus-based algorithm, 這個比較偏向統計,這個就有點像自然語言裡面,我們要去把一個grammar或者一個sentence
pattern,要找的話,一種方法當然是問語言學家有幾種講法,那現在比較新的話,就是譬如說把聯合報2003年,2004年的資料拿過來,然後跟你這個動詞有關的所有的這些sentence通通抓出來,然後看他是怎麼來使用的。所以這樣的時候,我可以把這個動詞,在任何時間點,我對哪個動詞有興趣,我甚至可以到google上面把所有的動詞抓出來,當然不是所有,就是選擇性的抓出來。抓出來到一個量後,你大約對他所有出現的情況都可以做某種程度的了解。那這樣的話,你就必須用到machine learning或者是knowledge-based的algorithm,所以這個在生物資訊上,也是一個蠻重要的技巧。所以,如果你是在做生物資訊的時候,要有一個概念,就是我們,eventually最好的生物資訊學家應該是生物背景出生的,然後他數學不錯,但這是一個dream。大學的時候就是雙學位,啊,就是生物和數學,不是生物和computer science,因為computer
science對我來說是too application-oriented,應該是生物和數學然後到研究所,他又學會programming如果他生物沒有丟掉,他可以去修更高等的bio-chemistry。
什麼是生物學家要的?什麼是computer science能協助他們的?:
你做出來的這些工具,事實上,是一定要在生物實驗室使用,沒有application的話這是沒有用的。在生物學的論文裡頭,conclusion是擺在很前面的,結論對他們來說是最重要的,你可以看到一個生物學的會議,那些生物學家埋頭翻,找論文的景象,他們看這些論文很快,因為他們只看結論。嗯,我們通常做出來的tool對生物學家講太rough,譬如我們做一個結構的predication,正確率90沒用95也沒用,所以他要100%。所以,我們給它的工具並不是說我們今天predict出來就拿去用,而是一個decision support。他可能對這一堆蛋白質都有興趣,那你告訴他,說這一百個蛋白質中間,我大概給你做一個結構的分類,哪一些是屬於這類,這類,這樣的話他就很容易知道說,雖然你不是100%,但是,你大概八九不離十,那我對中間某十個蛋白質,這類結構蛋白質會有興趣,這樣對他幫助其實非常大,而不是說他要exact solution。另外就是說,我們這個emphasis不是在這個解題的技巧有多fancy,多困難,多複雜,這是以前做computer science或做理論比較著重。現在是說,你解決生物的這個問題是不是真的很有用,對生物學家非常有用。
那第三個就是說,你能不能夠把這個工具和生物知識做某種程度的結合。像剛剛講的自然語言方面,如果你今天做生物資訊的literature mining的時候,你有沒有辦法把生物資訊或生物的knowledge儘量放在你的工具,放的越多越好,等下我們舉個例子就是我們在實驗室做的時候,也是有個困難,就是說knowledge表達的方式是以IT為主。所以今天你生物不同的知識要進來的時候,要怎麼樣參與在這個decision making process是個蠻困難的事情。然後最後corpus-based algorithm,我們一直強調這個,一般有些人用純粹machine learning,純粹machine
learning有一點困難就是說,它的predication受限於一般像分類的話在七八十左右,很難上的去。所以要有更多的knowledge把它補足才行,那我現在講三個簡單例子在我們實驗室做的比較多一點的。
第一個是protein secondary structure prediction,第二個是用核磁共振來決定蛋白質結構的assignment algorithm,這裡面各位生物不是很懂沒有關係我會大概用computer science的語言來講。第三個就是比較熟悉literature mining。那在protein
secondary structure prediction,這是發表在NAR 2004年的。
protein secondary structure prediction:
那protein structure我們剛剛從這個sequence開始,這是一條序列,20個做任意的排列,當然不是任意的,因為它有某程度上的distribution,但是我們不清楚。那secondary structure有點像是電話線,它本身會做某種程度的旋轉,或者是平板式的排列。然後,你這個電話線轉好了以後呢,這個轉好的線又會做tertiary structure,它又會有三度空間的排列旋轉。最後呢,是這些 structure,各位看這一塊,可以整個拿到這裡,有好幾塊這種 structure合起來會變成一個它像是四維的結構,這個四維並不是在四度空間,而是在另外一個層面的結合。那我們今天,這個secondary structure prediction,基本上是說,從這個primary structure,就是這個protein sequence怎麼樣去predict這個二維的這個結構。這個二維的結構大概分成三類,簡單的講起來分成三類,所以講起來是一個蠻簡單的問題。
這三類呢,一個是alpha-helix,就是這樣螺旋狀的,一個是beta-strand,平版狀,另外就是連接alpha-helix和beta-strand的中間,這些他們叫looks,所以基本的,主要的是alpha-helix和beta-strand這兩種。那secondary structure prediction就是說把一個蛋白質sequence,這是寫在上面,上面20個letter phrases,然後你把它轉成二維結構時只剩下三個letter,就是這上面寫的三個letter: L,E,H這三個 letter,怎樣把這三個 letter轉的越正確越好,這,當然這個有點像是注音輸入,對不對?例如你現在輸入蛋白質sequence,一輸入進去它就變成3個letter,這是你要的答案,這個原來上面可能是你的注音,下面是字,字是只有三種,非常簡單,但是,它依據不同的上下文它會轉,這個L,E,H會不一樣,所以呢,如果照以前做自然語言的這種的想法來看的話,這是一個translation的problem,就是說怎麼樣把translation做得很好,現在目前正確率差不多在80%,也就是說你大概80%的對應,大概會做對,其它的可能會做錯,那如果以自然語言角度,我們可以先去study它裡面有些什麼樣重要的word,phrases,很可惜,在protein sequence裡面,這個word跟phrases是undefined,它的結構跟我們一般自然語言是有蠻大的差距,它有的時候很妙的是說這個sequence,假設有1,2百個氨基酸,中間只要有一個做一點改變,他整個結構就完全翻掉,這有點像自然語言裡面寫的not,對不對?不,否定,類似這樣,就是這樣的一個情況你一個局部的,local的簡單的改變會引起整個三度空間的整個變換,這種現象在自然語言裡面是不太容易常常出現,偶爾會有。
那在protein裡面的話我們就希望說,這樣的情況下它的words,phrases到底是什麼?這是一個蠻有趣的問題,那當然,我們現在並沒有答案,我們今天做的方式就是,其實也是產生一個knowledge base,用這個knowledge base來做prediction,然後希望以後能夠放更多的,這些生物的知識進到這個knowledge base,這是以前的作法,statistical approaches,或是chemistry-based approaches,或是machine learning approaches。那這個目前最成功的是machine learning的這個approach,用neural net用的最多。那下面這點也是一個蠻有趣的,我們在自然語言裡面比較不容易有這個現象,protein sequence剛才我們講過有100個或1000個這個氨基酸排列出來,那我們可以把任兩個protein sequence做一個所謂的alignment,alignment就是說你去看這個它的一些氨基酸,可以對到一起,可以上下對到一起,然後哪些地方可以做deletion,insertion,substitution,這樣做完後,如果它的similarity,就是它對的,正確的那些氨基酸如果達到25%以上,這兩個結構就會similar,這個在自然語言裡面是沒有這個現象。你把兩個句子對應起來,他如果有25%的字可以對的好的話,就表示這兩個句子的意思是一樣,事實上不太可能,可是在這就是protein的一個很有趣的現象就是大部分到25%以上它的這個結構,它的三度空間結構算是它的meaning就已經完全固定下來,當然會有些許的boundary的差異,可是這大的差異是沒有的。
那這樣的話就是說,也給我們一個啟示就是,今天你要來處理這個所謂protein sequence的自然語言的話,你應該要採取一個什麼樣的態度,那我們今天,knowledge base的approach基本上處理是什麼呢?就是說,我們假設今天處理的protein sequence,它的similarity都蠻低的,都是25%以下,就sequence的similarity蠻低的,但是它有別的這個相似的東西,可以把它restore,所以我們最早做的一個方法就是說我去產生一些所謂的similar的比較小段的protein,譬如說你每7個做一個window size,然後這樣一直move下去,這會產生很多的這種小的序列,那這個序列是不是所有的可能都會出現也不見得,它的頻率分佈其實差異非常大,那我們這樣做的一個原因就是說,希望在這種所謂local的這些小段,每7個window裡面找到一些相似的,那這樣的話我們就可以組成一個7字詞的library,當然這並不是真正的protein的word,這是我們用自然語言作模擬,7字詞的這些小段,然後放在knowledge base裡面,那你來一個新的n node的protein的時候,我就去也是一樣7個window,7個window去move,看你可以找到knowledge base裡面哪些7字詞來跟你這個比較接近,然後,利用它們你已經有的結構,來做這個prediction。
OK,那這個global跟local就是我剛剛講的,其實這是一個蠻local的approach,你只是看那個7字的window啊,有沒有在knowledge base裡面,有沒有相對應的similarity,那global similarity事實上是看你說你這麼多7字詞,你這有百分之多少是跟我knowledge base裡面所存的詞,你看knowledge裡面把他想成一個字典一樣,你有多少詞可以對到字典裡面,如果對的不錯的話表示你的global的這個protein,跟我這個knowledge base也是蠻相似的。那這個細節我就不太詳述,就是怎麼樣產生這個7字詞的library,這是比較生物的knowledge,domain
knowledge多一點,所以,我們現在就假設knowledge
base裡面有很多的7字詞,所以你現在一個target
protein,這是我們要預設他結構的一個protein進來,剛才我講過,就是protein我們要把它轉成3個letter,就是H,E,L,然後希望正確率越高越好,然後做法上就是你現在每7個單元amine
acid就切一個window,這樣一直切下來,1,2,3,4,5,6,7,8,9,10,這樣一直切下來,然後,我每一個potential的這個wondow,都去knowledge
base裡面去做對應,看看有多少可以對到,那做對應的時候,你會發現,當然事實上我們最後轉的時候是對每一個amine
acid去轉,所以呢,我今天找到的knowledge
base的詞,只要能夠包含我這個位置的amine
acid,譬如說在這個x的位置啊我就把它抓進來,我這個potential可以投票的一個對象,所以,譬如說我這個x啊。這個knowledge
base裡面有很多詞嘛,那任何一個詞它只要包含我這個位置的話,我就把它抓過來做一個投票對象,所以這個紅色就是我現在有興趣的位置,那這些是我抓到這些在knowledge
base裡面的這些所謂的7字詞,然後呢,我去看一下到底這些7字詞裡面,因為knowledge
base裡面我們是假設它都已經轉好的,它的E,H,L,都已經轉好的,所以我都已經知道他最後是H,L,還是E,那我就去看這個字,它的這個位置是轉成H還是E還是L,所以用一個很簡單的一個
majority fold 的方式啊,如果它H最多的話啊,我就說我這個
n node的位置就是H,所以這個voting
score是很簡單的,那這個knowledge
base做完以後我們會有一個global的evaluation,這個match
rate是說我剛才這麼多window有多少,譬如說這個,一開始是1,2,3,4,5,6,7,這樣所有的window都產生,然後這些紅的是說他可以match到knowledge
base裡面的字典,這些的比例有多高,這就是我的match
rate,這算是一個比較global的evaluation,那我們現在我們根據這個實驗結果發現這個global
match rate如果到達80%以上,事實上,我們的knowledge
base的預測會比較準,knowledge
base預測就是我剛剛講的,用這些投票的方式,那這些80%以下,事實上它的這個信心值不夠高,這時我們就用一個現在最好的一個machine
learning的方法,譬如說side fact,用別人的prediction,80以上用我們的prediction,那這樣的一個結果呢,就可以improve。
這就是一個曲線,各位可以看到,這個是別人的prediction,是粉紅色的,這是用
nureral network的一個prediction,這個prediction跟你的這個match
rate幾乎是無關的,下面是說這個對我們knowledge
base來講這個n node sequence他可以match到的percentage,從10%,20%到80%,100,那可以看的到如果它的match的percantage越高的時候它的正確率,y軸是它的正確率,正確率越高,那另外一個用
neurual net 的方法對這個match rate的影響幾乎這很少,這個也是個優點,因為他在match
rate很低的時候就knowledge base可以提供的資訊很少的時候,它的正確率還是不錯,所以我們在...我們最後決定就是在80%啊,這個是一個cut
point,80以下我們就用它的neurual
net的方法,然後80%以上用我們的方法,這樣很快就可以得到一個improvement,而且這個improvement有一個好處就是你以後這個knowledge
base一定是越來越大,因為這個能夠被解出來的protein
structure,蛋白質結構越來越多,所以這個線,整體來會越來越往右移啊,停的越來越高,那這樣的情況下我們能夠match到比較好的80%以上的這些sequence的機會啊就越來越多,那這是我們最近做的一個improve,剛才那個缺點是什麼呢?剛才那個缺點就是說你前面這段是用別人的,後面這個可能你比人家好用。
你前面這一段是用別人的,後面這一段你可能比人家好的地方,那就用自己的.所以你的最後prediction rate百分之八十是別人的,百分之二十是自己的。那我們現在把這個方法,做一個某種程度的改變,讓我們是overload都比人家好,不是只有在前面用別人的,我們是在每一個位置幾乎比人家好的機率都滿高。所以現在這個大概是overload都比人家高兩個percent,以現在最好的方法能夠高兩個percent,這是前面knowledge base的方法。
核磁共振來決定蛋白質結構:
下面我們講一下,用核磁共振來決定這個蛋白質的結構的方法,這也是一個滿有趣的algorithm。這個是我們剛才畫過的primary structure,是一個sequence.然後呢,這個問題是要決定他三度空間的結構,tertiary structure。NMR是一個滿貴的儀器,是來 observe 這個 tertiary structure,他沒有像顯微鏡一樣可以看得到,他只能測一些所謂chemical的property,所以最後你可以得到一個瞎子摸象的結果,就是他剛才那個 NMR的機器,大概可以作十個不同的實驗,每個實驗可以 catch 到一部分的值.這些值是一些參數,他可以用來predict三度空間,或者決定三度空間結構的一個參數,所以說我們就等於有十個瞎子來看一個象,這個象就是真正的三度空間結構,每個瞎子都可以看到一部份。其實並不是三度空間結構,而是某些參數。你有沒有辦法能夠把這十種實驗的參數能夠混在一起,然後正確的決定三度空間結構。如果你要把這些combine起來的話,每個實驗他的參數都會有錯,這是很麻煩的。所以你需要是把這些實驗的值把他兜起來,兜起來做一個比較好的預測,這就是一個最後的結果。
Chemical Shift:
Chemical shift 這很難解釋,大家知道有這麼回事就是。這是中間某三個實驗可以測到的值。那比較重要的是說有一個實驗可以測到兩個點的值。另外一個實驗可以測到六個點,再來一個是四個點。這六個點就是兩個amine acid,一前一後,相關的一些值。那我們比較有興趣的是說,這是兩個點的實驗,實驗完你可以畫在這個 two dimensional plain上,他等於形成很多cluster.這些cluster,你有沒有辦法經過某種程度的過濾,把noise去掉,然後把有意義的結合起來。
剛才那個問題比較麻煩是說,原來我們想得很清楚的是說一個 amine acid可能有四個點,然後呢,相鄰的兩個 acid 會產生六個點,但是呢,你測到的實驗,他是沒有辦法分出來哪四個點是同一個實驗。就剛剛我講的瞎子摸象,他一個實驗,這些所有的四個點,他可能有一百個 observation全部混在一起,然後呢,另外一個實驗兩個點,他也全部混在一起。所以在這個時候,再加上他有 force positive,force negative的產生,你有沒有辦法把原來的每一個實驗的四個點和兩個點都能夠正確的抓出來。用一個例子來形容好了,假設你的朋友結婚,到一個飯店去吃飯,那門口有人可以幫你開車,他就幫你們一部一部車,依照次序,這是他開到旁邊的 garage,這個次序你當然是不知道的。但最後要散場的時候,他說很抱歉,今天那個電腦出了問題,你們手上雖然拿著票,但票的號碼跟次序是無關的。這個次序,現在完全get lost ,完全不知道,那我希望所有參加婚禮開車的人呢,你們出來站一排,看看說你前面大概是什麼車。比如說你大概是開 Camry,那你記得前面是一部 Volvo,後面也許是一部 Savrin,當然Volvo 有很多輛嘛,camry 也很多輛.所以現在就叫所有開車的人排隊,要重組 reverse engineer ,要把原來的 sequence 找出來.這個 sequence 原來是一個 protein 的 sequence,只是我剛剛瞎子摸象,我 observe 到的這些 parameter太多了,我現在把這些 parameter對到這些 protein sequence 上面,叫一個unminer acid.但是 unnemmer sequence 只有 20 個,像車子的號碼,車子的牌子一樣.假設這個是 verrory,有很多同樣的車,但是能不能藉由我剛才講的說,每個人大概記得說我前面是什麼車,後面是什麼車,然後這樣的組合,可以把他 disambiguous,次序是完全正確,這樣就可以把他這些車子還給各位,要不然的話,這個問題是不能解決的。那這裡面遇到的一個困難,像我剛剛講到的force positive,force negative,也許有人他說我沒開車,而他有開車,有人記性不好,他前面應該是camry他記成volve,這也是一個問題.那有人記錯了前面反而記成後面,那有人只記得前面不記得後面,這些都是我剛剛講的這些你要做這些reverse engineering遇到的困難。如果是最簡單的case,每個人的記性都非常好,記得都非常清楚,你還會遇到一個問題是說,camry前面是volve,後面是這個savrin的組合有好幾個,你還不知道是哪一輛,你還得要湊得很長之後才知道。所以類似的問題是你現在遇到的是這個生物資訊方面,把剛所謂的參數,要能對到 protein sequence上,所遇到的問題。那我大概講一下,你知道困難度就可以了。所以這是說,some people's memory is wrong, some just forget their neighbors,這樣的一個情況.這是以前的用的方法是 constrainted bipartile matching 的這個方法,但是他的問題有很多assumption還是不滿足。那我們現在用的一個是,我們用的是兩種方法,比較偏這個.我先讓你所有的可能都組合起來,就是剛剛講,比如說你前面是volve,後面是sarvin..沒關係,把你形成一個 possible 的 candidate,這 candidate 多到一定程度的時候很多competing的candidate,那我再利用一些 iterator 的方法,能夠把這個問題,一步一步解決。那其實比較困難的,不是剛才講說有很多 camry,很多 sarvin,很多車子都在裡面。如果你的記憶很清楚,這個perfect data 能夠做到百分之九十八以上,就可以做到把重組的那個正確率提高到百分之九十八。比較麻煩的是說有些 force negative,你記的不是很清楚,前面的車忘掉,會有人前後的車都忘掉,那這樣你怎麼把這個車的位置找出來,這是最困難的.所以我們這個作重組的過程中,有很多小的片段都組的不錯.這個時候,我們會用兩種方法,一種是maximum-independent set,互相沒有排斥性,不牴觸的這些sequence,排成一個比較更長的sequence,另外一個是generic algorithm.Generic algorithm有一個好處是,他會做某種combination,就是把一個可能是exponential growth的這種combination,這個正確率比較高的component或這些block能夠留下來,然後讓愈多正確的block這些sequence存活的機率愈高.到最後的話就是你存下來的那幾個中,有幾個是全部正確的.那剛才兩種方法,前面的那個independent set的paper是在今年recon 2005年被接受的,就是能後在兩組data上都有不錯效應,一個是91.4.各位想想看,以前的話他們真的就像我剛講的,幫我停車的那個boy,用紙牌去一張一張排,就是在實驗室,完全沒有電腦的輔助,把這整個sequence排出來,那它要排兩三個禮拜才排得完.現在我們的algorithm大概兩三秒就可以把百分之九十的問題解決掉,剩下來他就稍微排一下,因為還有一些生物的property它可以利用.所以一個是百分之九十,一個是百分之八三,就可以把這個問題,解決到相當不錯的程度.這就是我剛跟各位講的,一個decision-support system.當然我這個百分之九十也不知道是哪一個正確.不過他看一下,他大概就可以決定.所以到這個程度以上,對他的幫助就非常大.另外就是你遇到一個perfect data,這個是有error,就是有false positive,false negative蠻嚴重的.這個false positive可以到百分之五十以上,false negative也可能也到百分之五十以上.所以他不是我們一般能夠用discrete algorithm處理的這些問題.那遇到perfect data,如果你的ambiguity僅限於剛我們講說有很多camry的車,salvin的車,你前後的記憶都很清楚,很正確的話,可以做到百分之九八,這個是目前的方式.
literature mining:
最後簡單講一下literature mining.這個在生物學家裡面,我認為,以後資訊學家做的literature mining,這些algorithm解掉了就是解掉了,可能我講的三度空間這個NMR的這個結構,做出來的時候這個algorithm就結束了,不會有第二種可能.但是在literature mining這個問題,永遠就像google search一樣,你要search relation永遠層出不窮,變化無端.一般他們很有興趣的是說,今天我有一個疾病,那這些疾病跟哪一些基因有關係,你有沒有辦法很快.所以光是了解問題是沒有意義,而是要本身能把solution抓到,這個是最困難.所以這個disease,譬如哪一些基因跟高血壓有關.如果以後sequencing很快的話,每一個DNA sequence進去後,大概一個小時就全部出來,有什麼基因,在什麼地方都知道,那個時候就比較容易知道說,我患高血壓的機率是多高,就是genetically機率有多高.所以這一類問題,都是生物學家每天要問的問題。而且這個都非常expensive。
NER(named entities recognition):
那基礎的工作是named entities recognition,這個是比較自然語言上面的名詞,叫NER,NER其實就是說,你要把一個disease描述出來,或者把一個protein描述出來。你用google search或者是submain line這個abstrain search.他因為這些名稱都是生物學家自己寫的。
像我們今天,剛才講的out-timer disease,是用一個人的名字掛在一個病上面。所以一講到out-timer那個scenario就整個出來。但今天,protein有幾十萬幾百萬個,不可能再用人的名字去給一個protein的名字。所以今天protein是有點像 ... 各位看字典上面,也許你有一個牛津字典,另外一個是偽誓字典.同樣一個字在牛津字典有個一個解釋,在偽誓字典有個解釋對不對。
今天有點像說,寫在literature裡面的蛋白質不是用標準的ontology term,如果用標準time好了,semantic web盛行全世界,現在是沒有像這樣的事情,就是他用他的一個解釋的方式去描述這個字。這兩個字典,我今天要你做一個alignment,把牛津字典的所有解釋抓出來,不告訴你是哪一個字的解釋。然後你要align到這個偉博字典的interpretation,然後來看你的正確率,完全把中間dictionary entry全部拿掉,這個就是在生物裡面做NER。把protein name,DNA name要能後認出來他的困難度。另外假設你運氣不錯,可以把這些做到某個程度的話,下面才是輪到說,這兩個protein是不是有一個interaction。
我們今天剛講的,一個生化反應裡面,有很多protein和protein interaction。這個protein也許做修補工作,那個protein做切斷的工作。那你把這些protein之間的關係都找出來,在這個文句裡面,這個就是NER,兩個relation的recongition,document classification。譬如說,這個document,各位可以想,protein也許它有不同的function,你有沒有辦法,把這個文章裡面講的protein他的function做一個classification。另外一個,每一個不同文章內protein都做不同的classification。這就是一個完全在literature裡面做的一個mining。另外還有一些false positive,false negative是什麼意思呢,就有很多relation寫在文獻裡面,而他是錯的。因為他是根據生物實驗,譬如說我做個實驗裡面,我發現到一百個protein-protein interaction的possible candidate全部寫下來。那在生物實驗裡面他的正確率,事實上不到50%。我下次在重做的話沒有辦法repeat。那可能就有的relation是錯的,就不會在第二次出現。所以在這種情況,你不但是要找對relation,還要去看有多少paper提到跟這兩個protein有關係。也許只有這一篇提到的話,可能他的可信度就不夠高,或者另外一篇說這兩個是沒有關係的。那你這些文章都要蒐集過來,然後做一個比較,才能夠提供給所謂的生物學家或user一個比較所謂的confidence level,你對這個relation的confidence有多少。因為這些東西,他是最後要拿去畫他的paint sheet?,那譬如說有些domain他就非常困難,譬如說我們剛講說,假設是高血壓的基因,這篇文章裡面有沒有提到高血壓的基因?也許高血壓裡面有十幾種生化反應,他就提到說這兩個基因呢,會在某一種生化反應裡面,扮演一個很重要的角色。那事實上就是如果以這個生物學家來看,這個就是跟高血壓之病相關的。那這種事情的話,你沒有這個domain knowledge,你只是去看高血壓這個字,是完全沒有辦法達到這樣一個效果。所以呢,這個lecture mining,其實裡面複雜的問題很多。如果你今天要開一個這個新的google公司,我想開到生物資訊裡面,應該生意會比較好一點,所以基本上,biology need a search engine than GOOGLE。那Named Entity Recognition,在自然語言裡面就有點像人名地名機構名。人名並不是簡單的三個字,他可能是新竹市衛生局局長某某某,整個合起來才算一個Named Entity。所以他難的地方就在於是不是能夠把每一個item都知道他不同的效果,然後可以把這整個entity抓到,這是relation的地方,譬如說這句話,"The ZAP-70 mutant studied here could be phosphorylated on tyrosine when associated to the TCR zeta chain and was able to bind p56(lck)",這裡面你可以抓到什麼樣的relation。有的時候,他會寫一些簡寫,譬如PI。PI是Permeability Index和另外一個完全不是p和i開頭的,這個是生物學家用語,你要把這些簡稱抓到。所以現在可能有三種pi「註」,是完全不一樣的。那你抓到之後還要知道translation,還要知道他是哪一個,這等於是他的翻譯一樣,所以這變得很重要。那剛才我們同樣一個句子,就ZAP-70 mutant,這裡面就會抓到一個relation,這relation就是說,這個ZAP-70 entity就會bind p56(lck),事實上這句話是最後才出現的。was able to bind ...,那中間講一大堆其他的話,你有沒有辦法能夠過濾掉。那一般data mining,就說純粹做database data mining的話,就會把這些co-location抓出來,ZAP-70是另外一個data chain。事實上這裡面呢,並不是主要的。然後把這些抓起來,就說他有某些關係,事實上,這錯誤的機率(false negative)太高了,false positive很高。
這就是剛才講的named entity抓出來以後,他的disambiguation到底是哪一個。現在事實上有些做data mining的公司,專門在做這一部份。他就是把這個paper裡面所提到的基因,他不是說這個基因是哪個基因,他說這個基因是整個document把他classified成某一個list或answer(某一種基因)。所以在literature mining,我現在提到的只是基礎(Named Entity Recognition),先把所有蛋白質,DNA RNA名詞可以找對,這個就非常困難。然後找對以後呢,把他的relation抓出來,因為下面是我們要做的生化反應結構的預測。
Naming template:
那這裡面要用到的方法,naming template。template有兩種,一個是蛋白質的名稱事實上是有的是用他的function來描述,所以是一個phrase,那這個phrase組成的方式就是某種template,第二種是dictionary based,其實效果是有限的。我們在web上面,你用dictionary去search很多東西,有很多新的詞,事實上不再dictionary裡面,也是一個大的問題。machine learning這些方法都可以用在named entity recognition上面。那我們現在用的比較多的是maximum entropy比和CRF。現在用的training set是日本的一個機構做的。training set他的一個很大的缺點是他20%是錯的,他的標示不consistent,就是所有的這種做training set tech corpus的一個問題。那如果你說不用這個tech corpus,你到網路上去抓的話,那個noise太大,所以這也是一個很大的困難。在自然語言現在很多強調不用dictionary,然後自動產生dictionary,自動產生翻譯。然後在這個基因裡面,就是蛋白質名詞的對應上面,因為他牽涉domain knowledge太多,所以這是一個很困難,像他構詞上面的features,POS的features,以及semantec trigger features,hand-noun features,這些都需要做很深入的研究。Morphological Features結尾是cin mide的話他們是什麼。這只是一個大概,不見得一定要照這個。Head-noun,他裡面有這樣的一個名詞的話,可能是什麼樣的一個東西,他可以one-gram two-gram (bi-gram),所以他一個protein名稱可能有四個字,你中間如果有出現這個的話是蛋白質的機率多高,DNA的機率多高。orthographic features這些是很無聊的一些詞,在protein名稱常出現一個括弧或出現一個點,你也要把他變成一個feature,然後你才能做machine learning。那做relation recognition的話最好要有一個robust partial parser而不要full parser,full parser蠻困難的,然後藥用很多的template還有統計的方法,在運用co-occurrence relation,所以大概就簡單介紹到這些。
相關資料:
蛋白質構造:
像其它巨分子一樣,蛋白質鏈也是由小分子單位 (胺基酸) 一個一個連接成的。
▲1.1 一級構造:
探究蛋白質構造可由胺基酸序列開始,循序依其複雜度分成四個層次。
a.蛋白質的骨架:
蛋白質的長條胺基酸序列,是為其 一級構造 (primary structure)。 此一級構造的一端為 N-端 (-NH2),另一端為 C-端 (-COOH),而以 H2N-C-C-[N-C-C]x-N-C-COOH 為骨架,其中黑體字 C 代表各胺基酸單位的 a 碳。
b.一級構造信息是由 DNA 所決定:
一級構造是蛋白質最終構形的根本,各級構造的訊息都決定於胺基酸的序列;而胺基酸序列是根據 DNA 的核苷酸序列轉譯而來,故最終的信息是存留於細胞核的核酸中。
c.蛋白質序列可由核酸序列得知:
探討一級構造主要在分析該蛋白質之 胺基酸序列,近來胺基酸序列多由該蛋白質的 cDNA 經分子群殖 (cloning) 篩選出,再經核酸定序後轉譯成胺基酸而來。
d.有三個官能基的胺基酸:
除了構造之外,一段固定的胺基酸序列可能有某種特定的生理功能,稱之為 signal peptide (信息序列),同一 signal 可以在許多不同的蛋白質分子上重複出現。 例如蛋白質在 C-端若有 Lys-Asp-Glu-Leu (KDEL) 的序列,則會被回收到內質網去。
▲1.2二級構造:
蛋白質長鏈捲繞成堅固而規則的二級構造,是其構形的基礎單位。
胜鍵 的雙鍵性質形成 胜鍵平面;又因兩α碳上的R基團與前後相鄰基團的引力或斥力,使得兩相鄰胜鍵平面間的轉動,限制在一定角度範圍,而造成規律的兩種主要構形: α螺旋 (a helix)、β 長帶 (β sheet),是為 二級構造 (secondary structure),它們的主要構成力量都是 氫鍵。 氫鍵在細胞內的角色不但重要,而且分佈非常廣泛。
a.α螺旋:
每 3.6 個胺基酸捲繞一圈,成為右手旋的螺旋構造,遇 Pro 則中止;由相鄰兩胜鍵平面所夾的角度,可以預測 a 螺旋或其它二級構造的生成 (Ramachandran Plot)。 分子內氫鍵可在螺旋骨架間加上支架,更使得 α螺旋成為圓筒狀,有堅固的構形,也是三級構造的組成單位之一。 肌紅蛋白 由八段長短不等的 a 螺旋所組成。
b.β長帶:
像彩帶般的構形,多由數條彩帶組合而成,相鄰彩帶之間以 氫鍵 接合,編成一片堅固的盾形平面。 依相鄰彩帶的 N→C 方向關係,可分為 同向 (parallel) 及 逆向 (antiparallel) 兩大類。β長帶多由 R 基團較小的胺基酸 (如 Ala, Gly, Ser) 組成。
c. 其它螺旋構造:
除了 α螺旋外,蛋白質也可以其它方式摺疊成螺旋構造,但每一單位螺旋的胺基酸數目不同。 當連續有數個 Pro (如 PVPAPIPP) 時,會捲成稱為 polyproline 的螺旋,每三個胺基酸轉一圈,橫切面是一個正三角形。
a. Turn 轉折:
連接α螺旋或β長帶時,胜肽鏈需做劇烈的轉折,以接近 180 度的方式摺返,這些轉折點稱為 turns;其中βturn 由四個胺基酸組成 (多含有 Gly); γturn 則由三個胺基組成,且由一個 Pro 造成主要轉折。 Turns 多分佈在蛋白質的表面,也很容易誘生抗體。
b. 不規則形:
除上述構造外,尚有構形不規則的連結片段,稱為 不規則形;而在蛋白質分子兩個端點附近的胜肽,活動性較大,形狀經常變化,則為 任意形 (random coil)。
▲
大部分蛋白質的三級構造捲繞成 球狀 (globular),已是有特定構形的活性分子。
a. 二級鍵組合三級構造:
分子內各部分的二級構造再相互組合,構成完整球形的三級構造 (tertiary structure);其構成的作用力有 離子鍵、氫鍵、疏水鍵、金屬離子 等作用力;其中疏水鍵對水溶性蛋白質三級構造之穩定性,貢獻最大。
b. 疏水鍵穩定蛋白質核心:
水溶性蛋白質的核心緊密,多由疏水性胺基酸組成。 由於疏水性胺基酸為外界水環境所排斥,可以穩定地包埋在分子內部,維持蛋白質的完整三級構造。
c. 雙硫鍵加強構造:
蛋白質分子內兩個 Cys 上的 -SH 可經氧化而成 雙硫鍵 (-S-S-),雙硫鍵可加強蛋白質的立體構造。 在細胞中,雙硫鍵的形成可能需要靠酵素的催化。
a. Domain 的形成:
某些二級構造經常會聚在一起,例如 αααα, βαβ或 α8β8 筒狀構造等,都可自形成一小區域,稱為 domain (功能區塊),若干 domains 再組成一完整蛋白質的三級構造。 小的蛋白質通常只有一個 domain,較大蛋白質則含有兩個以上的 domains。 經常重複出現的 domain,可能有特定功用,可視為一種二級構造的再現單位 (motif)。
b. 蛋白質構形的資訊儲藏在胺基酸序列中:
建構蛋白質最終立體構形的藍圖,其資訊是貯藏在一級構造的胺基酸序列中;因此某些蛋白質 (如 RNase) 在 變性 後,仍然可以回復原來的立體構形 (原態 native),稱為 復性。
c. Chaperone 幫助摺疊正確構形:
並非所有的變性蛋白質都可如 RNase 般復性回原態,細胞內有一類巨大分子稱為 chaperone,可以幫助蛋白質正確地摺疊成原態分子。
d. 以胺基酸序列只能正確預測二級構造:
若已知某蛋白質的胺基酸序列,則可以電腦運算預知某段是
α螺旋、β長帶或是轉折等二級構造,相當準確。
但更複雜的三級立體構造,目前則較不容易預測準確。
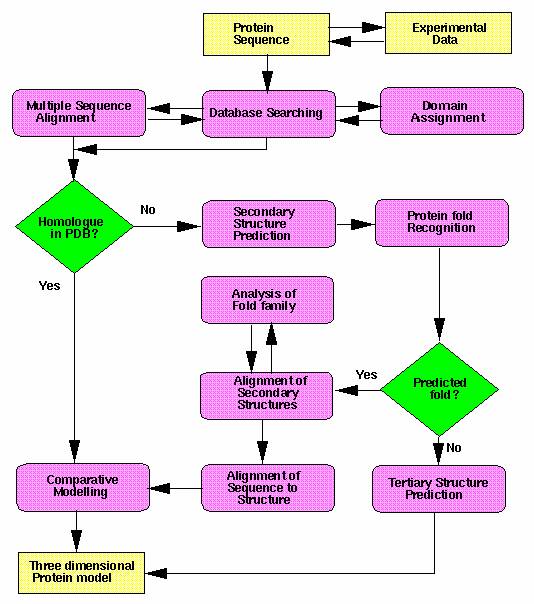
蛋白質構造的預測方法流程圖:
a. 蛋白質再經修飾:
很多蛋白質的三級構造,即為獨立而具有活性的分子; 但有些則要再加上 輔脢、輔因子 (如 金屬離子) 或 輔基 (prosthetic group, 如 肌紅蛋白 中的 heme);有些則要再修飾以糖分子成為 醣蛋白 (glycoprotein);脂蛋白 (lipoprotein) 要連接脂質;更有的要再與其它相同或不同的蛋白質分子結合,形成四級構造 (如 血紅蛋白)。
b. 有些蛋白質以裂解產生活性:
有些蛋白質的胺基酸鏈要先經過切斷或刪除某段胜肽後,才能有活性 (如 胰島素)。胰島素是由一條基因轉譯出來的,切成三段後,其中兩段以雙硫鍵結合成具活性的三級構造,並非四級構造;這是蛋白質活性的調控方式之一。
▲
蛋白質再聚合成四級構造,可調節控制其功能,或組成巨大構造分子。
a. 次體組成四級構造:
若數個相同或不同的三級構造分子,再結合成一較大的複合體,才能進行完整的活性功能,則稱為 四級構造 (quaternary structure)。 構成四級構造的每一單位分子,稱為次體 (subunit);通常各次體之間無共價結合,而以二級鍵為主要結合力量。每一條次體蛋白質,都是由一個完整基因所轉譯出來的。
b. 四級構造之目的:
若四級構造的任一次體與受質結合之後,會誘導增強其它次體與受質之結合能力,而加速反應,則稱為正協同作用 (positive cooperativity),反之則為 負協同作用。 四級構造在 調控 蛋白質的活性上非常重要,許多酵素是重要的例子;但典型具四級構造,且有複雜調控機制的血紅蛋白並非酵素。
c. 構成性四級構造:
許多病毒的蛋白質外殼,具有規律而巨大的四級構造組成。
蛋白質研究技術:
蛋白質性質與構造檢定:
a. 蛋白質定量法: 染料 Coomassie Blue 與蛋白質結合後,會由褐色變為藍色; 由反應前後藍色吸光度的改變,與已知蛋白質的標準曲線比較,即可推知樣本中蛋白質的濃度;現在一般多使用 Bradford's
method。
b.
分子量測定法: 可利用 膠體過濾法、超高速離心法
(ultracentrifugation)、或 膠體電泳法
(gel electrophoresis) 來測定蛋白質的原態分子量;SDS
膠體電泳
則可測定次體分子量。 電泳
同時可以檢定蛋白質的純度如何,是解析力極佳的分析工具。
c.
等電點
(pI): 等電焦集法
(isoelectric focusing) 極類似膠體電泳,但可測得蛋白質的等電點。 等電點是蛋白質帶電性質的重要指標,當環境的 pH 等於其等電點時,此蛋白質的淨電荷為零;大於其等電點時,淨電荷為負,反之則為正電荷。
d.
胺基酸組成: 蛋白質以鹽酸水解成游離胺基酸,再以分析各種胺基酸之含量。
e.
蛋白質立體構造: 以
X 光繞射法 分析蛋白質結晶,可計算出其分子的細微立體構造;近來也流行應用 核磁共振法 (NMR) 測定水溶液中蛋白質的立體構造。
f.
質譜分析: 目前的質譜儀分析技術,已經能夠處理分子量較大的蛋白質,則可以定出蛋白質的精確分子量;甚至可以檢查該蛋白質所產生的各個片段,推出其胺基酸序列。
資料來源: http://juang.bst.ntu.edu.tw/BCbasics/Protein1.htm
相關網站:
1. www.bioisland.com.cn/guihua/kjgh/html/4.htm
在生物島規劃--科技發展規劃中提到了智能算法研究、設計及計算能行性和複雜性理論和超級數據計算處理中心。
有一些關於中國863計劃的信息,由於863計劃中也涉及到一些算法的研究。
5. http://www.ebi.ac.uk/dos/clustalw/
可以下載Clustalw多序列聯配程序。
6. http://www.isrec.isb-sib.ch/
可以了解一些關於BOXSHADE程序的信息。
上面有一些關於相鄰片斷組裝程序的算法描述。
8. http://gnomic.stanford.edu/
上有GenScan程序的算法。
上有Genie程序的算法描述。
10. http://genome.cbs.dtu.dk/
上有ECOPARSE的程序算法描述。
11.http://cbil.humgen.upenn.edu/
上有GeneParser這個關於基於動態規劃方法的基因識別程序的算法。
12.ftp://iubio.bio.indiana.edu
上有關於SeqPup的程序。